Zodostat
- ENG
- မြန်မာ

For the use of Registered Medical Practitioner or a Hospital or Laboratory
Daprodustat Tablets 2 mg
ZODOSTAT 2
COMPOSITION
Each film-coated tablet contains:
Daprodustat ——— 2 mg
Daprodustat Tablets 4 mg
ZODOSTAT 4
COMPOSITION
Each film-coated tablet contains:
Daprodustat ——— 4 mg
Daprodustat Tablets 6 mg
ZODOSTAT 6
COMPOSITION
Each film-coated tablet contains:
Daprodustat ——— 6 mg
1. DESCRIPTION
ZODOSTAT contains daprodustat, an inhibitor of hypoxia inducible factor (HIF), prolyl 4- hydroxylases (PH)1, PH2 and PH3. The chemical name of daprodustat is N-[(1,3-dicyclohexylhexahydro-2,4,6-trioxopyrimidin-5-yl) carbonyl] glycine. The molecular formula of daprodustat is CH,N,O, and its molecular mass is 393.43. The structural formula is shown below.
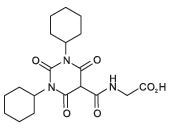
Daprodustat is a white to off-white powder that is poorly soluble in water.
Each ZODOSTAT oral tablet contains 1 mg, 2 mg, 4 mg or 6 mg of daprodustat.
2. CLINICAL PHARMACOLOGY
2.1 Mechanism of Action
Daprodustat is a reversible inhibitor of HIF-PH1, PH2 and PH3 (IC50 in the low nM range). This activity results in the stabilization and nuclear accumulation of HIF-1a and HIF-20 transcription factors, leading to increased transcription of the HIF-responsive genes, including erythropoietin.
2.2 Pharmacodynamics
Effects on Erythropoiesis
Daprodustat increases endogenous erythropoietin in a dose-dependent manner within 6 to 8 hours after administration. With repeat doses, peak increases in reticulocyte counts occur between 7 and 15 days, with subsequent increases in red blood cell production. New hemoglobin steady-state levels are reached several weeks (approximately 4 weeks in ESA-users and approximately 16-20 weeks in ESA-non-users) after initial administration.
Effects on Iron Metabolism and Utilization
Daprodustat increased serum transferrin and total iron binding capacity (TIBC) and decreased serum ferritin, transferrin saturation, and hepcidin when administered for 52 weeks in adults on dialysis with anemia due to CKD.
Cardiac Electrophysiology
At a dose 10 times the maximum recommended dose, daprodustat does not prolong the QTc interval to any clinically relevant extent.
2.3 Pharmacokinetics
Daprodustat exposure generally increases in a dose-proportional manner over the range of approved doses. Steady-state concentrations are achieved within 24-hours of dosing.
Absorption
Following oral administration, daprodustat is readily absorbed with median time to peak concentration (T) in healthy subjects ranging from 1 hour to 4 hours. The absolute bioavailability of daprodustat is 65%. Administration of ZODOSTAT with a high fat/high calorie meal did not significantly alter daprodustat exposure compared to administration in the fasted state.
Distribution
Daprodustat has an approximately equal distribution between plasma and blood cells (blood:plasma ratio of 1.23). Following intravenous dosing, the volume of distribution at steady- state in healthy subjects is 14.3 L. In vitro, plasma protein binding of daprodustat is >99%.
Elimination
The terminal elimination half-life of daprodustat is approximately 1 hour to 4 hours.
Metabolism: In vitro, daprodustat is primarily metabolized by CYP2C8 (95% contribution), with a minor contribution by CYP3A4 (5%).
Following oral or intravenous administration of radiolabeled daprodustat to healthy adults, approximately 40% of the total circulating radioactivity in plasma was daprodustat, and the remaining 60% was metabolites.
In patients treated with ZODOSTAT, the parent drug is the principal circulating component in plasma. Three metabolites, each accounting for more than 10% of circulating drug-related material, have been identified; in vitro and non-clinical data suggest that each may contribute to the pharmacollogic response in vivo; however, the extent of this contribution is unknown.
Excretion: Mean clearance from plasma was 18.9 L/h, which correlates to blood clearance of 15 L/h and equates to a hepatic extraction of approximately 18%.
Within seven days of an oral dose of radiolabeled daprodustat, 74% of the radioactivity was recovered in the feces, and 21% in the urine. Approximately 99.5% of the dose was excreted as oxidative metabolites, with the rest accounted for by daprodustat.
Specific Populations
Elderly: Population pharmacokinetic analyses in adults with CKD (22 years to 93 years) showed that age did not influence the pharmacokinetics of daprodustat.
Renal Impairment: The steady-state exposure of daprodustat is similar in patients with normal renal function and those with varying degrees of renal impairment; daprodustat exposure is not significantly impacted by hemodialysis or peritoneal dialysis. The systemic exposure of daprodustat metabolites was higher in. patients with Stage 3 to 5 CKD compared to those with normal renal function. Exposures of metabolites were higher on non-dialysis days compared to dialysis days.
Hepatic Impairment: Following administration of a single
ZODOSTAT 6 mg dose, mean daprodustat Cmax and AUC increased by 2-fold and unbound exposure increased by 2.3-fold in subjects with moderate hepatic impairment (Child-Pugh Class B) compared to subjects with normal hepatic and renal function, For those with mild hepatic impairment (Child-Pugh Class A), mean daprodustat Cmax was similar while AUC increased by 1.5-fold and unbound Cmax and AUC increased by 1.6 and 2.2-fold, respectively, compared to subjects with normal hepatic and renal function. The effect of severe hepatic impairment (Child-Pugh Class C) on the pharmacokinetics of daprodustat is unknown as there have been no studies of ZODOSTAT in patients with severe hepatic impairment.
3. INDICATIONS AND USAGE
ZODOSTAT is indicated for the treatment of anemia due to chronic kidney disease (CKD) in adults who have been receiving dialysis for at least four months.
Limitations of Use
ZODOSTAT has not been shown to improve quality of life, fatigue, or patient well-being. ZODOSTAT is not indicated for use:
– As a substitute for red blood cell transfusions in patients who require immediate correction of anemia.
– For treatment of anemia of chronic kidney disease in patients who are not on dialysis.
4. DOSAGE AND ADMINISTRATION
4.1 Pre-Treatment and On-Treatment Evaluations of Anemia, Iron Stores, and Liver Tests
Evaluation of Anemia and Iron Stores
Before initiating ZODOSTAT, correct and exclude other causes of anemia (e.g., vitamin deficiency, metabolic or chronic inflammatory conditions, bleeding). Evaluate the iron status in all patients before and during treatment with ZODOSTAT. Administer supplemental iron therapy when serum ferritin is less than 100 mcg/mL or when serum transferrin saturation is less than 20%. The majority of patients with CKD will require supplemental iron during the course of therapy.
Liver Testing
Assess serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and total bilirubin prior to initiation of ZODOSTAT. Repeat the liver tests if the patient develops signs or symptoms that could be consistent with liver disease during treatment with ZODOSTAT.
4.2 Important Dosing Information
Individualize dosing and use the lowest dose of ZODOSTAT sufficient to reduce the need for red blood cell transfusions, Do not target a hemoglobin higher than 11 g/dL..
ZODOSTAT can be taken with or without food, and without regard to concomitant administration of iron or phosphate binders.
ZODOSTAT should be swallowed whole. Tablets should not be cut, crushed, or chewed.
ZODOSTAT can be administered without regard to the timing or type of diallysis.
If a dose of ZODOSTAT is missed, it should be taken as soon as possible, unless it is the same day as the next dose. In this case, the missed dose should be skipped, and the next dose taken at the usual time. Double-doses should not be taken to make-up for a missed dose.
4.3 Recommended Starting Dose of ZODOSTAT
Adults with Anemia Due to Chronic Kidney Disease Receiving Dialysis for at least 4 Months
Adults Not Being Treated with an ESA: For adults not being treated with an ESA, the starting dose of ZODOSTAT is based on the hemoglobin level (see Table 1). Dose modifications are needed for patients receiving concomitant treatment with a moderate CYP2C8 inhibitor or moderate hepatic impairment.
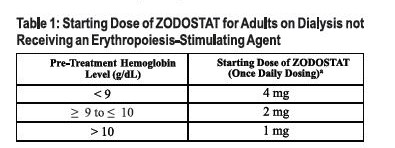
See dosing modifications if the patient has moderate hepatic impairment and if the patient is on a moderate CYP2C8 inhibitor.
Adults Being Switched from an ESA: For adults being switched from an ESA to ZODOSTAT, the starting dose of ZODOSTAT is based on the dose regimen of the ESA at the time of substitution (see Table 2). Dose modifications are needed for patients receiving concomitant treatment with a moderate CYP2C8 inhibitor or moderate hepatic impairment.
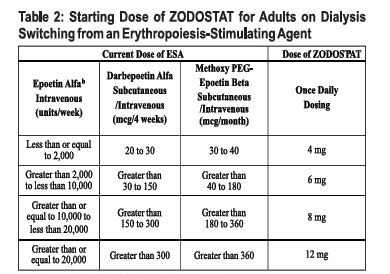
ESA-Erythropoiesis stimulating agent.
* See dosing modifications if the patient has moderate hepatic impairment and if the patient is on a moderate CYP2C8 inhibitor.
For patients on subcutaneous epoetin alfa, convert the epoetin alfa subcutaneous dose to intravenous dose equivalent by multiplying the subcutaneous dose received per week by 1.42 to obtain the weekly intravenous dose.
4.4 Monitoring Response to Therapy and Dose Adjustment
Following initiation of therapy and after each dose adjustment, monitor hemoglobin every 2 weeks for the first month and then every 4 weeks thereafter.
When adjusting doses of ZODOSTAT, consider hemoglobin rate of rise, rate of decline and hemoglobin variability. Do not increase the dose of ZODOSTAT more frequently than once every 4 weeks.
– If the dose of ZODOSTAT needs to be adjusted, increase or decrease by one dose level at a time (see Table 3).
– Decrease the dose of ZODOSTAT if hemoglobin increases rapidly (e.g., greater than 1 g/dL over 2 weeks or greater than 2 g/dL over 4 weeks) or if the hemoglobin exceeds 11 g/dL.
– If hemoglobin exceeds 12 g/dL, interrupt treatment with ZODOSTAT. When the hemoglobin is within the target range, treatment may be restarted at one dose level lower (see Table 3).
– Treatment with ZODOSTAT should not be continued beyond 24 weeks of therapy if a clinically meaningful increase in hemoglobin level is not achieved. Alternative explanations for an inadequate response should be sought and treated before re-starting therapy.

4.5 Dosage Modification for Hepatic Impairment
Reduce the starting dose of ZODOSTAT by half (see Tables 1 and 2) in patients with moderate hepatic impairment (Child-Pugh Class B) except in patients whose starting dose is already 1 mg.
Use of ZODOSTAT in patients with severe hepatic impairment (Child-Pugh Class C) is not recommended.
4.6 Dosage Modification for Concomitant Treatment with Moderate CYP2C8 Inhibitors
Reduce the starting dose of ZODOSTAT by half (see Tables 1 and 2) in patients who are on clopidogrel or a moderate CYP2C8 inhibitor except in patients whose starting dose is already 1 mg.
Monitor hemoglobin and adjust the dose of ZODOSTAT when initiating or stopping therapy with clopidogrel or a moderate CYP2C8 inhibitor during treatment with ZODOSTAT.
5. DRUG INTERACTIONS
5.1 CYP2C8 Inhibitors
Concomitant administration of strong CYP2C8 inhibitors (e.g., gemfibrozil) with ZODOSTAT is contraindicated due to a marked increase in daprodustat exposure.
Concomitant administration of moderate CYP2C8 inhibitors (e.g.. clopidogrel) increases daprodustat exposure. Reduce the starting dose of ZODOSTAT by half when initiating treatment in patients on clopidogrel or a moderate CYP2C8 inhibitor except in patients whose starting dose is already 1 mg. Monitor hemoglobin and adjust the dose of ZODOSTAT when initiating or stopping therapy with clopidogrel or a moderate CYP2C8 inhibitor during treatment with ZODOSTAT.
5.2 CYP2C8 Inducers
CYP2C8 inducers (e.g., rifampin) may decrease daprodustat exposure, which may result in loss of efficacy, Monitor hemoglobin and adjust the dose of ZODOSTAT when initiating or stopping therapy with CYP2C8 inducers during treatment with.
6. CONTRAINDICATIONS
– ZODOSTAT is contraindicated in patients:
– Receiving a strong CYP2C8 inhibitor such as gemfibrozil
– With uncontrolled hypertension
7. WARNINGS AND PRECAUTIONS
7.1 Increased Risk of Death, Myocardial Infarction, Stroke, Venous Thromboembolism, and Thrombosis of Vascular Access
ZODOSTAT increases the risk of arterial and venous thrombotic events, that may be fatal, including myocardial infarction, stroke, venous thromboembolism and vascular access thrombosis. Patients with cardiovascular or cerebrovascular disease are at increased risk of these events. Avoid use in patients with a history of myocardial infarction, cerebrovascular event, or acute coronary syndrome within the 3 months prior to starting ZODOSTAT.
A rate of hemoglobin rise of greater than 1 g/dL over 2 weeks may contribute to these risks. Targeting a hemoglobin level of greater than 11 g/dL is expected to further increase the risk of death and arterial and venous thrombotic events, as occurs with ESAs, which also increase erythropoietin levels.
No trial has identified a hemoglobin target level, dose of ZODOSTAT, or dosing strategy that does not increase these risks. Use the lowest dose of ZODOSTAT sufficient to reduce the need for red blood transfusions. Adherence to dosing and hemoglobin monitoring recommendations is important to avoid excessive erythropoiesis.
Advise patients to seek immediate medical attention if they develop signs or symptoms of myocardial infarction, stroke, venous thromboembolism, or thrombosis of vascular access, Evaluate and manage promptly if these occur.
7.2 Risk of Hospitalization for Heart Failure
Consider the patient’s history of heart failure when deciding whether to prescribe ZODOSTAT. Advise patients of the symptoms and signs of heart failure and to immediately report any worsening to their healthcare provider.
7.3 Hypertension
ZODOSTAT is contraindicated in patients with uncontrolled hypertension, Cases of hypertensive crisis including hypertensive encephalopathy and seizures have also been reported in patients receiving ZODOSTAT. Periodically monitor blood pressure and adjust or initiate anti-hypertensive therapy as needed.
7.4 Gastrointestinal Erosion
Advise patients of the symptoms and signs of gastric and esophageal erosions and of gastrointestinal bleeding and to seek prompt medical care if these occur, Consider this risk particularly in patients at increased risk for gastrointestinal erosions, such as those with a history of gastrointestinal erosion, peptic ulcer disease, use of concomitant medications that increase the risk of gastrointestinal erosion, and current tobacco smokers and alcohol drinkers.
7.5 Serious Adverse Events in Patients with Anemia Due to Chronic Kidney Disease and Not on Dialysis
The safety of ZODOSTAT has not been established for the treatment of anemia due to CKD in adults not on dialysis and its use is not recommended in this setting.
In a large cardiovascular outcomes trial in adults with anemia of CKD who were not on dialysis (ASCEND-ND), an increased risk of cardiovascular mortality, stroke, thromboembolism, serious acute kidney injury, hospitalization for heart failure, and serious gastrointestinal erosions was observed in patients treated with ZODOSTAT compared to rhEPO.
7.6 Malignancy
Because increased hypoxia inducible factor (HIF)-1 levels may be associated with unfavorable effects on cancer growth, ZODOSTAT has not been studied and is not recommended in patients with active malignancies. No evidence of increased carcinogenicity was observed in animal studies.
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data with ZODOSTAT use in pregnant women are insufficient to establish a drug associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. There are risks to the mother and the fetus associated with CKD.
Daprodustat administered orally to pregnant rats and rabbits during the period of organogenesis was associated with adverse fetal outcomes, including embryonic and fetal loss and reduced fetal weight, at doses that caused maternal toxicity and polycythemia. Advise pregnant women of the potential risk to the fetus.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk: CKD in pregnancy increases the risk for maternal hypertension, preeclampsia, miscarriage, stillbirth, preterm delivery, low birth weight infants, and polyhydramnios.
8.2 Lactation
Risk Summary
There are no data on the presence of daprodustat in human milk, the effects on the breastfed child, or the effects on milk production. Daprodustat is present in the milk of lactating rats. When a drug is present in animal milk, it is likely that the drug will be present in human milk. Given the serious adverse reactions seen in adults treated with ZODOSTAT, such as thrombotic vascular events, advise patients not to breastfeed during treatment with ZODOSTAT, and for one week after the final dose.
8.3 Pediatric Use
Safety and effectiveness of ZODOSTAT in pediatric patients have not been established.
8.4 Geriatric Use
Of the total number of subjects treated with ZODOSTAT in the ASCEND-D study (n = 2,964), 480 (32%) subjects were aged 65 years and older, and 159 (11%) were aged 75 years and older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, No other reported clinical experience has identified differences in responses between the elderly and younger patients.
8.5 Hepatic Impairment
No adjustment of the starting dose is required in patients with mild hepatic impairment (Child-Pugh Class A). Reduce the starting dose of ZODOSTAT by half in patients with moderate hepatic impairment (Child-Pugh Class B) except in patients whose starting dose is already 1 mg. ZODOSTAT has not been studied in patients with severe hepatic impairment (Child-Pugh Class C). Therefore, ZODOSTAT is not recommended in patients with severe hepatic impairment.
9. ADVERSE REACTIONS
– Increased Risk of Death, Myocardial Infarction, Stroke, Venous Thromboembolism, and Thrombosis of Vascular Access
– Risk of Hospitalization for Heart Failure
– Hypertension
– Gastrointestinal Erosion
10. DRUG ABUSE AND DEPENDENCE
10.1 Controlled Substance
ZODOSTAT contains daprodustat, which is not a controlled substance.
10.2 Abuse
Drug abuse is intentional non-therapeutic use of a drug, even once, for its rewarding psychological or physiological effects. Abuse of ZODOSTAT may be seen in athletes for the effects on erythropoiesis.
Abuse-Related Adverse Reactions
There are no data on the abuse of ZODOSTAT in humans. Daprodustat and its metabolites neither selectively penetrate the central nervous system, nor produce behavioral effects in animals that are consistent with central nervous system activity. Misuse of drugs that increase erythropoiesis, such as ZODOSTAT, by healthy persons may lead to polycythemia, which may be associated with life-threatening cardiovascular complications (e.g., stroke, myocardial infarction, and thromboembolism).
11. OVERDOSAGE
Headache and gastrointestinal adverse reactions (e.g., nausea) may be seen with acute overdose with ZODOSTAT, There is no specific antidote. Hemodialysis will not substantially remove daprodustat because it is highly protein bound.
12. STORAGE
Store below 30°C.
13. PRESENTATION
Alu-Alu Blister pack of 3 x 10’s in a carton along with pack insert.
KEEP MEDICINE OUT OF REACH OF CHILDREN.
Product of:
Zifam Pinnacle Pty. Ltd.,
Sydney, Australia.
Manufactured by:
Zifam Pyrex Myanmar Co. Ltd.,
Lot C6, Zone A, Thilawa SEZ, Thanlyin & Kyauk Tan Township, Yangon, Myanmar.