



Grace Imatinib
- ENG
- မြန်မာ
For the use only of a registered medical practitioner or a hospital or a laboratory Rx
Grace Imatinib
Imatinib Tablets
COMPOSITION
Imatinib 400mg Film-coated Tablets
Each Film-coated Tablet contains Imatinib Mesylate equivalent to Imatinib 400 mg
Excipients q.s.
PHARMACEUTICAL FORM
Film-coated Tablet.
THERAPEUTICINDICATIONS
imatinib is indicated for the treatment of
– adult and paediatric patients with newly diagnosed Philadelphia chromosome (bcr-abl) positive (Ph+) chronic myeloid leukaemia (CML) for whom bone marrow transplantation is not considered as the first line of treatment.
– adult and paediatric patients with Ph+CML in chronic phase after failure of interferon-alpha therapy, or in accelerated phase or blast crisis.
– adult and paediatric patients with newly diagnosed Philadelphia chromosome positive acute lymphoblastic leukaemia (Ph+ALL) integrated with chemotherapy.
– adult patients with relapsed or refractory Ph+ALL as monotherapy.
– adult patients with myelodysplastic/myeloproliferative diseases (MDS/MPD) associated with platelet-derived growth factor receptor (PDGFR) gene re-arrangements.
– adult patients with advanced hypereosinophilic syndrome (HES) and/or chronic eosinophilic leukaemia (CEL) with FIP1L1-PDGFRa rearrangement.
The effect of Imatinib on the outcome of bone marrow transplantation has not been determined.
Imatinib is indicated for
– the treatment of adult patients with Kit (CD 117) positive unresectable and/or metastatic malignant gastrointestinal stromal tumours (GIST).
– the adjuvant treatment of adult patients who are at significant risk of relapse following resection of Kit (CD117)-positive GIST. Patients who have a low or very low risk of recurrence should not receive adjuvant treatment.
– the treatment of adult patients with unresectable dermatofibrosarcoma protuberans (DFSP) and adult patients with recurrent and/or metastatic DFSP who are not eligible for surgery.
The effect of imatinib on the outcome of bone marrow transplantation has not been determined.
In adult and paediatric patients, the effectiveness of Imatinib is based on overall haematological and cytogenetic response rates and progression-free survival in CML, on haematological and cytogenetic response rates in Ph+ALL, MDS/MPD, on haematological response rates in HES/CEL and on objective response rates in adult patients with unresectable and/or metastatic GIST and DFSP and on recurrence-free survival in adjuvant GIST. The experience with Imatinib in patients with MDS/MPD associated with PDGFR gene re-arrangements is very limited. Except in newly diagnosed chronic phase CML, there are no controlled trials demonstrating a clinical benefit or increased survival for these diseases.
POSOLOGY AND METHOD OF ADMINISTRATION
Therapy should be initiated by a physician experienced in the treatment of patients with haematological malignancies and malignant sarcomas, as appropriate.
For doses other than 400 mg and 800 mg. a 100 mg divisible tablet is available.
The prescribed dose should be administered orally with a meal and a large glass of water to minimize the risk of gastrointestinal irritations. Doses of 400 mg or 600 mg should be administered once daily, whereas a daily dose of 800 mg should be administered as 400 mg twice a day, in the morning and in the evening.
For patients unable to swallow the film-coated tablets, the tablets may be dispersed in a glass of still water or apple juice. The required number of tablets should be placed in the appropriate volume of beverage (approximately 50 ml for a 100 mg tablet, and 200 ml for a 400 mg tablet) and stirred with a spoon. The suspension should be administered immediately after complete disintegration of the tablet(s)
Posology for CML in adult patients
The recommended dosage of Imatinib is 400 mg/day for adult patients in chronic phase CML. Chronic phase CML is defined when all of the following criteria are met blasts < 15% in blood and bone marrow, peripheral blood basophils <20%, platelets > 100 x 109/1.
The recommended dosage of Imatinib is 600 mg/day for adult patients in accelerated phase. Accelerated phase is defined by the presence of any of the following: blasts≥15% but <30% in blood or bone marrow, blasts plus promyelocytes≥30% in blood or bone marrow (providing <30% blasts), peripheral blood basophils≥20%, platelets <100 x 109/1 unrelated to therapy.
The recommended dose of Imatinib is 600 mg/day for adult patients in blast crisis. Blast crisis is defined as blasts ≥30% in blood or bone marrow or extramedullary disease other than hepatosplenomegaly.
Treatment duration: In clinical trials, treatment with Imatinib was continued until disease progression. The effect of stopping treatment after the achievement of a complete cytogenetic response has not been investigated.
Dose increases from 400 mg to 600 mg or 800 mg in patients with chronic phase disease, or from 600 mg to a maximum of 800 mg (given as 400 mg twice daily) in patients with accelerated phase or blast crisis may be considered in the absence of severe adverse drug reaction and severe non- leukaemia-related neutropenia or thrombocytopenia in the following circumstances: disease progression (at any time), failure to achieve a satisfactory haematological response after at least 3 months of treatment, failure to achieve a cytogenetic response after 12 months of treatment, or loss of a previously achieved haematological and/or cytogenetic response. Patients should be monitored closely following dose escalation given the potential for an increased incidence of adverse reactions at higher dosages.
Posology for CML in children
Dosing for children should be on the basis of body surface area (mg/m²). The dose of 340 mg/m² daily is recommended for children with chronic phase CML and advanced phase CML (not to exceed the total dose of 800 mg). Treatment can be given as a once daily dose or alternatively the daily dose may be split into two administrations one in the morning and one in the evening. The dose recommendation is currently based on a small number of paediatric patients. There is no experience with the treatment of children below 2 years of age.
Dose increases from 340 mg/m² daily to 570 mg/m² daily (not to exceed the total dose of 800 mg) may be considered in children in the absence of severe adverse drug reaction and severe non-leukaemia- related neutropenia or thrombocytopenia in the following circumstances: disease progression (at any time); failure to achieve a satisfactory haematological response after at least 3 months of treatment, failure to achieve a cytogenetic response after 12 months of treatment, or loss of a previously achieved haematological and/or cytogenetic response. Patients should be monitored closely following dose escalation given the potential for an increased incidence of adverse reactions at higher dosages.
Posology for Ph+ALL in adult patients
The recommended dose of Imatinib is 600 mg/day for adult patients with Ph+ ALL. Haematological experts in the management of this disease should supervise the therapy throughout all phases of care.
Treatment schedule: On the basis of the existing data, Imatinib has been shown to be effective and safe when administered at 600 mg/day in combination with chemotherapy in the induction phase, the consolidation and maintenance phases of chemotherapy for adult patients with newly diagnosed Ph+ALL. The duration of Imatinib therapy can vary with the treatment programme selected, but generally longer exposures to Imatinib have yielded better results.
For adult patients with relapsed or refractory Ph+ALL Imatinib monotherapy at 600 mg/day is safe, effective and can be given until disease progression occurs.
Posology for Ph+ALL in children
Dosing for children should be on the basis of body surface area (mg/m²). The dose of 340 mg/m² daily is recommended for children with Ph+ALL (not to exceed the total dose of 600 mg).
Posology for MDS/MPD
The recommended dose of Imatinib is 400 mg/day for adult patients with MDS/MPD
Treatment duration. In the only clinical trial performed up to now, treatment with Imatinib was continued until disease progression. At the time of analysis, the treatment duration was a median of 47 months (24 days-60 months).
Posology for HES/CEL
The recommended dose of Imatinib is mg/day for adult patients with HES/CEL.
Dose increase from 100 mg to 400 mg may be considered in the absence of adverse drug reactions if assessments demonstrate an insufficient response to therapy.
Treatment should be continued as long as the patient continues to benefit.
Posology for GIST
The recommended dose of Imatinib is 400 mg/day for adult patients with unrespectable and/or metastatic malignant GIST.
Limited data exist on the effect of dose increases from 400 mg to 600 mg or 800 mg in patients progressing at the lower dose.
Treatment duration: In clinical trials in GIST patients, treatment with Imatinib was continued until disease progression. At the time of analysis, the treatment duration was a median of 7 months (7 days to 13 months). The effect of stopping treatment after achieving a response has not been investigated.
The recommended dose of Imatinib is 400 mg/day for the adjuvant treatment of adult patients following resection of GIST. Optimal treatment duration is not yet established. Length of treatment in the clinical trial supporting this indication was 36 months.
Posology for DFSP
The recommended dose of Imatinib is 800 mg/day for adult patients with DFSP.
Dose adjustment for adverse reactions
Non-haematological adverse reactions
If a severe non-haematological adverse reaction develops with Imatinib use, treatment must be withheld until the event has resolved. Thereafter, treatment can be resumed as appropriate depending on the initial severity of the event.
If elevations in bilirubin > 3 x institutional upper limit of normal (IULN) or in liver transaminases > 5 x IULN occur, Imatinib should be withheld until bilirubin levels have returned to < 1.5 x IULN and transaminase levels to <2.5 x IULN. Treatment with Imatinib may then be continued at a reduced daily dose. In adults the dose should be reduced from 400 to 300 mg or from 600 to 400 mg, or from 800 mg to 600 mg, and in children from 340 to 260 mg/m2/day.
Haematological adverse reactions
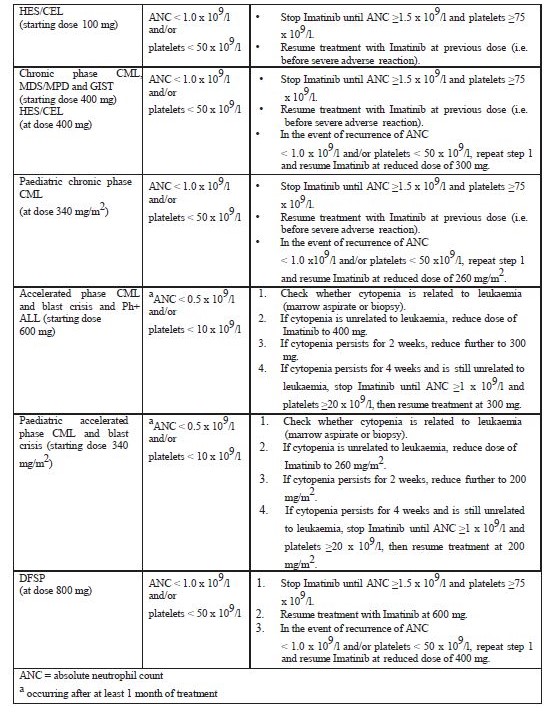
Dose reduction or treatment interruption for severe neutropenia and thrombocytopenia are recommended as indicated in the table below.
Dose adjustments for neutropenia and thrombocytopenia.
Special populations
Paediatric use: There is no experience in children with CML below 2 years of age and with Ph+ALL below 1 year of age. There is very limited experience in children with MDS/MPD, DFSP, GIST and HES/CEL
The safety and efficacy of imatinib in children with MDS/MPD, DFSP, GIST and HES/CEL aged less than 18 years of age have not been established in clinical trials. Currently available published data are summarized but no recommendation on a posology can be made.
Hepatic insufficiency: Imatinib is mainly metabolised through the liver. Patients with mild, moderate or severe liver dysfunction should be given the minimum recommended dose of 400 mg daily.
The dose can be reduced if not tolerated.
Liver dysfunction classification:
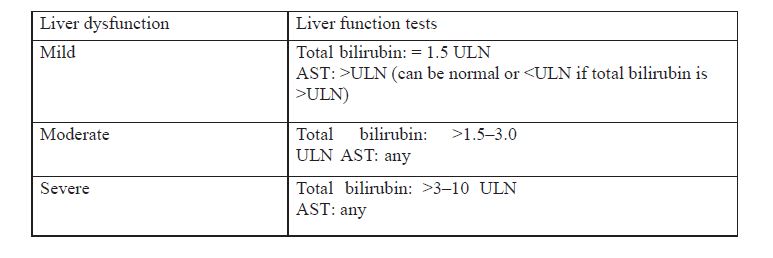
ULN-upper limit of normal for the institution
AST-aspartate aminotransferase
Renal insufficiency: Patients with renal dysfunction or on dialysis should be given the minimum recommended dose of 400 mg daily as starting dose. However, in these patients caution is recommended. The dose can be reduced if not tolerated. If tolerated, the dose can be increased for lack of efficacy.
Older people: Imatinib pharmacokinetics have not been specifically studied in older people. No significant age-related pharmacokinetic differences have been observed in adult patients in clinical trials which included over 20% of patients age 65 and older. No specific dose recommendation is necessary in older people.
CONTRAINDICATIONS
Hypersensitivity to the active substance or to any of the excipients listed.
WARNINGS AND PRECAUTIONS
When Imatinib is co-administered with other medicinal products, there is a potential for drug interactions. Caution should be used when taking Imatinib with protease inhibitors, azole antifungals, certain macrolides, CYP3A4 substrates with a narrow therapeutic window (e.g. cyclosporine, pimozide, tacrolimus, sirolimus, ergotamine, diergotamine, fentanyl, alfentanil, terfenadine, bortezomib, docetaxel, quinidine) or warfarin and other coumarin derivatives.
Concomitant use of imatinib and medicinal products that induce CYP3A4 (e.g. dexamethasone, phenytoin, carbamazepine, rifampicin, phenobarbital or Hypericum perforatum, also known as St. John’s Wort) may significantly reduce exposure to Imatinib, potentially increasing the risk of therapeutic failure. Therefore, concomitant use of strong CYP3A4 inducers and imatinib should be avoided.
Hypothyroidism
Clinical cases of hypothyroidism have been reported in thyroidectomy patients undergoing levothyroxine replacement during treatment with Imatinib. Thyroid-stimulating hormone (TSH) levels should be closely monitored in such patients.
Hepatotoxicity
Metabolism of Imatinib is mainly hepatic, and only 13% of excretion is through the kidneys. In patients with hepatic dysfunction (mild, moderate or severe), peripheral blood counts and liver enzymes should be carefully monitored. It should be noted that GIST patients may have hepatic metastases which could lead to hepatic impairment.
Cases of liver injury, including hepatic failure and hepatic necrosis, have been observed with imatinib. When imatinib is combined with high dose chemotherapy regimens, an increase in serious hepatic reactions has been detected. Hepatic function should be carefully monitored in circumstances where imatinib is combined with chemotherapy regimens also known to be associated with hepatic dysfunction.
Fluid retention
Occurrences of severe fluid retention (pleural effusion, oedema, pulmonary oedema, ascites, and superficial oedema) have been reported in approximately 2.5% of newly diagnosed CML patients taking Imatinib. Therefore, it is highly recommended that patients be weighed regularly. An unexpected rapid weight gain should be carefully investigated and if necessary appropriate supportive care and therapeutic measures should be undertaken. In clinical trials, there was an increased incidence of these events in older people and those with a prior history of cardiac disease. Therefore, caution should be be exe exercised in patients with cardiac dysfunction.
Patients with cardiac disease
Patients with cardiac disease, risk factors for cardiac failure or history of renal failure should be monitored carefully, and any patient with signs or symptoms consistent with cardiac or renal failure should be evaluated and treated.
In patients with hypereosinophilic syndrome (HES) with occult infiltration of HES cells within the myocardium, isolated cases of cardiogenic shock/left ventricular dysfunction have been associated with HES cell degranulation upon the initiation of imatinib therapy. The condition was reported to be reversible with the administration of systemic steroids, circulatory support measures and temporarily withholding imatinib. As cardiac adverse events have been reported uncommonly with imatinib, a careful assessment of the benefit/risk of imatinib therapy should be considered in the HES/CEL population before treatment initiation.
Myelodysplastic/ myeloproliferative diseases with PDGFR gene re-arrangements could be associated with high eosinophil levels. Evaluation by a cardiology specialist, performance of an echocardiogram and determination of serum troponin should therefore be considered in patients with HES/CEL, and in patients with MDS/MPD associated with high eosinophil levels before imatinib is administered. If either is abnormal, follow-up with a cardiology specialist and the prophylactic use of systemic steroids (1-2 mg/kg) for one to two weeks concomitantly with imatinib should be considered at the initiation of therapy.
Gastrointestinal haemorrhage
In the study in patients with unresectable and/or metastatic GIST, both gastrointestinal and intra- tumoural haemorrhages were reported. Based on the available data, no predisposing factors (eg tumour size, tumour location, coagulation disorders) have been identified that place patients with GIST at a higher risk of either type of haemorrhage. Since increased vascularity and propensity for bleeding is a part of the nature and clinical course of GIST, standard practices and procedures for the monitoring and management of haemorrhage in all patients should be applied.
In addition, gastric antral vascular ectasia (GAVE), a rare cause of gastrointestinal haemorrhage, has been reported in post-marketing experience in patients with CML, ALL and other diseases. When needed, discontinuation of Imatinib treatment may be considered.
Tumour lysis syndrome
Due to the possible occurrence of tumour lysis syndrome (TLS), correction of clinically significant dehydration and treatment of high uric acid levels are recommended prior to initiation of Imatinib.
Hepatitis B reactivation
Reactivation of hepatitis B in patients who are chronic carriers of this virus has occurred after these patients received BCR-ABL tyrosine kinase inhibitors. Some cases resulted in acute hepatic failure or fulminant hepatitis leading to liver transplantation or a fatal outcome.
Patients should be tested for HBV infection before initiating treatment with Imatinib. Experts in liver disease and in the treatment of hepatitis B should be consulted before treatment is initiated in patients with positive hepatitis B serology (including those with active disease) and for patients who test positive for HBV infection during treatment. Carriers of HBV who require treatment with Imatinib should be closely monitored for signs and symptoms of active HBV infection throughout therapy and for several months following termination of therapy.
Phototoxicity
Exposure to direct sunlight should be avoided or minimised due to the risk of phototoxicity associated with imatinib treatment. Patients should be instructed to use measures such as protective clothing and sunscreen with high sun protection factor (SPF).
Laboratory tests
Complete blood counts must be performed regularly during therapy with Imatinib. Treatment of CML patients with Imatinib has been associated with neutropenia or thrombocytopenia. However, the occurrence of these cytopenias is likely to be related to the stage of the disease being treated and they were more frequent in patients with accelerated phase CML or blast crisis as compared to patients with chronic phase CML. Treatment with Imatinib may be interrupted or the dose may be reduced.
Liver function (transaminases, bilirubin, alkaline phosphatase) should be monitored regularly in patients receiving Imatinib.
In patients with impaired renal function, imatinib plasma exposure seems to be higher than that in patients with normal renal function, probably due to an elevated plasma level of alpha-acid glycoprotein (AGP), an imatinib-binding protein, in these patients. Patients with renal impairment should be given the minimum starting dose. Patients with severe renal impairment should be treated with caution. The dose can be reduced if not tolerated.
Long-term treatment with imatinib may be associated with a clinically significant decline in renal function. Renal function should, therefore, be evaluated prior to the start of imatinib therapy and closely monitored during therapy, with particular attention to those patients exhibiting risk factors for renal dysfunction. If renal dysfunction is observed, appropriate management and treatment should be prescribed in accordance with standard treatment guidelines.
Paediatric population
There have been case reports of growth retardation occurring in children and pre-adolescents receiving imatinib. The long-term effects of prolonged treatment with Imatinib on growth in children are unknown. Therefore, close monitoring of growth in children under Imatinib treatment is recommended.
Fertility, pregnancy and lactation
Women of childbearing potential
Women of childbearing potential must be advised to use effective contraception during treatment.
Pregnancy
There are limited data on the use of imatinib in pregnant women. There have been post-marketing reports of spontaneous abortions and infant congenital anomalies from women who have taken Imatinib. Studies in animals have however shown reproductive toxicity and the potential risk for the foetus is unknown. Imatinib should not be used during pregnancy unless clearly necessary. If it is used during pregnancy, the patient must be informed of the potential risk to the foetus.
Breast-feeding
There is limited information on imatinib distribution on human milk. Studies in two breast-feeding women revealed that both imatinib and its active metabolite can be distributed into human milk.
The milk plasma ratio studied in a single patient was determined to be 0.5 for imatinib and 0.9 for the metabolite, suggesting greater distribution of the metabolite into the milk. Considering the combined concentration of imatinib and the metabolite and the maximum daily milk intake by infants, the total exposure would be expected to be low (~10% of a therapeutic dose). However, since the effects of low-dose exposure of the infant to Imatinib are unknown, women taking imatinib should not breast-feed.
Fertility
In non-clinical studies, the fertility of male and female rats was not affected. Studies on patients receiving Imatinib and its effect on fertility and gametogenesis have not been performed. Patients concerned about their fertility on Imatinib treatment should consult with their physician.
Effects on ability to drive and use machines
Patients should be advised that they may experience undesirable effects such as dizziness, blurred vision or somnolence during treatment with imatinib. Therefore, caution should be recommended when driving a car or operating machinery.
OVERDOSAGE
Experience with doses higher than the recommended therapeutic dose is limited. Isolated cases of Imatinib overdose have been reported spontaneously and in the literature. In the event of overdose the patient should be observed and appropriate symptomatic treatment given. Generally the reported outcome in these cases was “improved” or “recovered”. Events that have been reported at different dose ranges are as follows:
Adult population
1200 to 1600 mg (duration varying between 1 to 10 days): Nausea, vomiting, diarrhoea, rash, erythema, oedema, swelling, fatigue, muscle spasms, thrombocytopenia, pancytopenia, abdominal pain, headache, decreased appetite.
1800 to 3200 mg (as high as 3200 mg daily for 6 days): Weakness, myalgia, increased creatine phosphokinase, increased bilirubin, gastrointestinal pain.
6400 mg (single dose). One case reported in the literature of one patient who experienced nausea, vomiting, abdominal pain, pyrexia, facial swelling, decreased neutrophil count, increased transaminases.
8 to 10 g (single dose): Vomiting and gastrointestinal pain have been reported.
Paediatric population
One 3-year-old male exposed to a single dose of 400 mg experienced vomiting, diarrhoea and anorexia and another 3-year-old male exposed to a single dose of 980 mg experienced decreased white blood cell count and diarrhea.
In the event of overdose, the patient should be observed and appropriate supportive treatment given.
PHARMACOLOGICAL PROPERTIES
Mechanism of Action
Pharmacotherapeutic group: protein-tyrosine kinase inhibitor, ATC code: L01XE01
Imatinib is a small molecule protein-tyrosine kinase inhibitor that potently inhibits the activity of the Ber-Abl tyrosine kinase (TK), as well as several receptor TKs. Kit, the receptor for stem cell factor (SCF) coded for by the c-Kit proto-oncogene, the discoidin domain receptors (DDR1 and DDR2), the colony stimulating factor receptor (CSF-1R) and the platelet-derived growth factor receptors alpha and beta (PDGFR-alpha and PDGFR-beta). Imatinib can also inhibit cellular events mediated by activation of these receptor kinases.
Pharmacodynamics
Imatinib is a protein-tyrosine kinase inhibitor which potently inhibits the Bcr-Abl tyrosine kinase at the in vitro, cellular and in vivo levels. The compound selectively inhibits proliferation and induces apoptosis in Ber-Abl positive cell lines as well as fresh leukaemic cells from Philadelphia chromosome positive CML and acute lymphoblastic leukaemia (ALL) patients. vivo the compound shows anti-tumour activity as a single agent in animal models using Ber-Abl positive tumour cells.
In Imatinib is also an inhibitor of the receptor tyrosine kinases for platelet-derived growth factor (PDGF), PDGF-R, and stem cell factor (SCF), c-Kit, and inhibits PDGF-and SCF-mediated cellular events. In vitro, imatinib inhibits proliferation and induces apoptosis in gastrointestinal stromal tumour (GIST) cells, which express an activating kit mutation. Constitutive activation of the PDGF receptor or the Abl protein tyrosine kinases as a consequence of fusion to diverse partner proteins or constitutive production of PDGF have been implicated in the pathogenesis of MDS/MPD, HES/CEL and DFSP. Imatinib inhibits signalling and proliferation of cells driven by dysregulated PDGFR and Abl kinase activity.
Clinical studies in chronic myeloid leukaemia
The effectiveness of Imatinib is based on overall haematological and cytogenetic response rates and progression-free survival. Except in newly diagnosed chronic phase CML, there are no controlled trials demonstrating a clinical benefit, such as improvement in disease-related symptoms or increased survival.
Three large, international, open-label, non-controlled phase II studies were conducted in patients with Philadelphia chromosome positive (Ph+) CML in advanced, blast or accelerated phase disease, other Ph+ leukaemias or with CML in the chronic phase but failing prior interferon-alpha (IFN) therapy. One large, open-label, multicentre, international randomised phase III study has been conducted in patients with newly diagnosed Ph+ CML. In addition, children have been treated in two phase I studies and one phase II study.
In all clinical studies 38-40% of patients were≥60 years of age and 10-12% of patients were≥70 years of age.
Chronic phase, newly diagnosed: This phase III study in adult patients compared treatment with either single-agent Imatinib or a combination of interferon-alpha (IFN) plus cytarabine (Ara-C) Patients showing lack of response (lack of complete haematological response (CHR) at 6 months, increasing WBC, no major cytogenetic response (MCyR) at 24 months), loss of response (loss of CHR or MCYR) or severe intolerance to treatment were allowed to cross over to the alternative treatment arm. In the Imatinib arm, patients were treated with 400 mg daily. In the IFN arm, patients were treated with a target dose of IFN of 5 MIU/m²/day subcutaneously in combination with subcutaneous Ara-C 20 mg/m2/day for 10 days/month.
A total of 1,106 patients were randomised, 553 to each arm. Baseline characteristics were well balanced between the two arms. Median age was 51 years (range 18-70 years), with 21.9% of patients > 60 years of age. There were 59% males and 41% females, 89.9% caucasian and 4.7% black patients. Seven years after the last patient had been recruited, the median duration of first-line treatment was 82 and 8 months in the Imatinib and IFN arms, respectively. The median duration of second-line treatment with Imatinib was 64 months. Overall, in patients receiving first-line Imatinib, the average daily dose delivered was 406±76 mg. The primary efficacy endpoint of the study is progression-free survival.
Progression was defined as any of the following events: progression to accelerated phase or blast crisis, death, loss of CHR or MCYR, or in patients not achieving a CHR an increasing WBC despite appropriate therapeutic management. Major cytogenetic response, haematological response, molecular response (evaluation of minimal residual disease), time to accelerated phase or blast crisis and survival are main secondary endpoints. Response data are shown in Table 2.
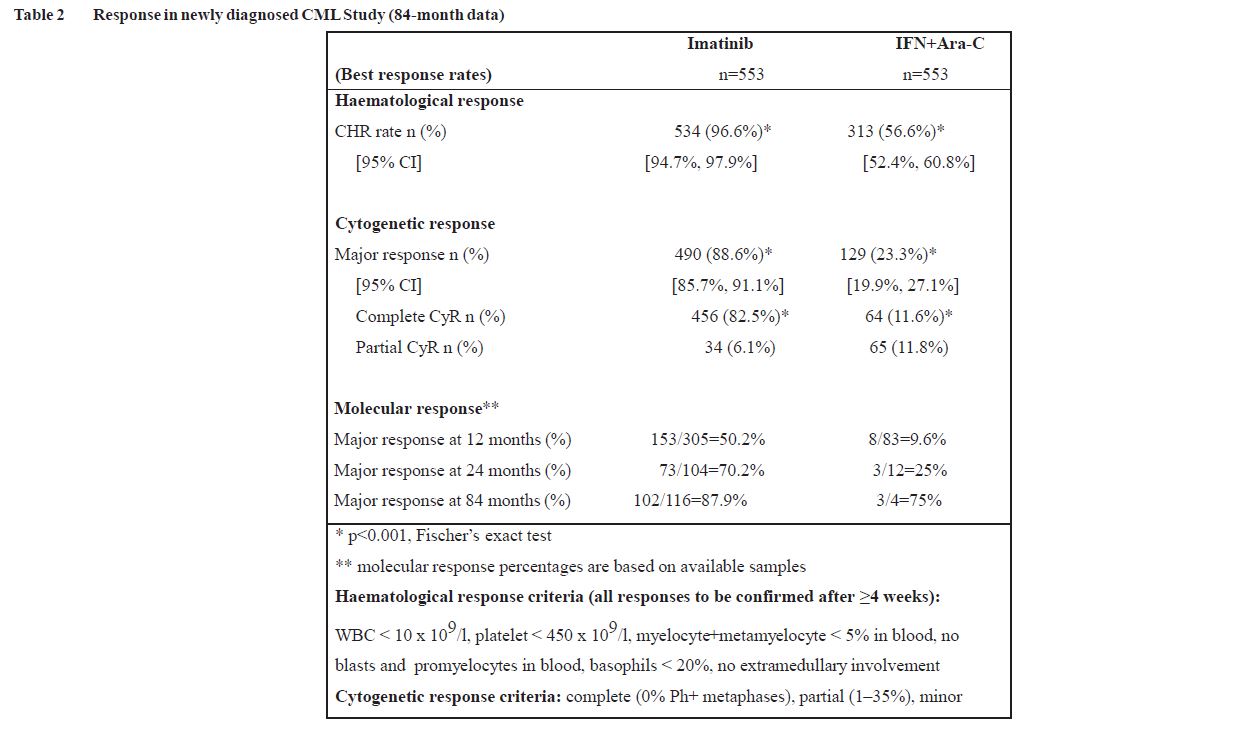
Rates of complete haematological response, major cytogenetic response and complete cytogenetic response on first-line treatment were estimated using the Kaplan-Meier approach, for which non-responses were censored at the date of last examination. Using this approach, the estimated cumulative response rates for first-line treatment with Imatinib improved from 12 months of therapy to 84 months of therapy as follows. CHR from 96.4% to 98.4% and CCyR from 69.5% to 87.2%, respectively.
With 7 years follow-up, there were 93 (16.8%) progression events in the Imatinib arm: 37 (6.7%) involving progression to accelerated phase/blast crisis, 31 (5.6%) loss of MCyR, 15 (2.7%) loss of CHR or increase in WBC, and 10 (1.8%) CML unrelated deaths. In contrast, there were 165 (29.8%) events in the IFN+Ara-Carm, of which 130 occurred during first-line treatment with IFN+Ara-C The estimated rate of patients free of progression to accelerated phase or blast crisis at 84 months was significantly higher in the Imatinib arm compared to the IFN arm (92.5% versus 85.1%, p<0.001). The annual rate of progression to accelerated phase or blast crisis decreased with time on therapy and was less than 1% annually in the fourth and fifth years. The estimated rate of progression-free survival at 84 months was 81.2% in the Imatinib arm and 60.6% in the control arm p < 0.001 ). The yearly rates of progression of any type for Imatinib also decreased over time.
A total of 71 (12.8%) and 85 (15.4%) patients died in the Imatinib and IFN+Ara-C groups, respectively. At 84 months the estimated overall survival is 86.4% (83, 90) vs. 83.3% (80, 87) in the randomised Imatinib and the IFN+Ara-C groups, respectively (p=0.073, log-rank test). This time-to-event endpoint is strongly affected by the high crossover rate from IFN+Ara-C to Imatinib. The effect of Imatinib treatment on survival in chronic phase, newly diagnosed CML has been further examined in a retrospective analysis of the above reported Imatinib data with the primary data from another Phase III study using IFN+Ara-C (n = 325) in an identical regimen. In this retrospective analysis, the superiority of Imatinib over IFN+Ara-C in overall survival was demonstrated (p < 0.001) within 42 months, 47 (8.5%) Imatinib patients 63 (19.4%) IFN+Ara-C patients had died.
The degree of cytogenetic response and molecular response had a clear effect on long-term outcomes in patients on Imatinib. Whereas an estimated 96% (93%) of patients with CCyR (PCYR) at 12 months were free of progression to accelerated phase/blast crisis at 84 months, only 81% of patients without MCyR at 12 months were free of progression to advanced CML at 84 months p < 0.001 overall, p = 0.25 between CCyR and PCYR). For patients with reduction in Ber-Abl transcripts of at least 3 logarithms at 12 months, the probability of remaining free from progression to accelerated phase/was 99% at 84 months. Similar findings were found based on a 18-months landmark analysis.
In this study, dose escalations were allowed from 400 mg daily to 600 mg daily, then from 600 mg daily to 800 mg daily. After 42 months of follow-up, 11 patients experienced a confirmed loss (within 4 weeks) of their cytogenetic response. Of these 11 patients, 4 patients escalated up to 800 mg daily, 2 of whom regained a cytogenetic response (1 partial and 1 complete, the latter also achieving a molecular response), while of the 7 patients who did not escalate the dose, only one regained a complete cytogenetic response. The percentage of some adverse reactions was higher in the 40 patients in whom the dose was increased to 800 mg daily compared to the population of patients before dose increase (n = 551) The more frequent adverse reactions included gastrointestinal haemorrhages, conjunctivitis and elevation of transaminases or bilirubin. Other adverse reactions were reported with lower or equal frequency.
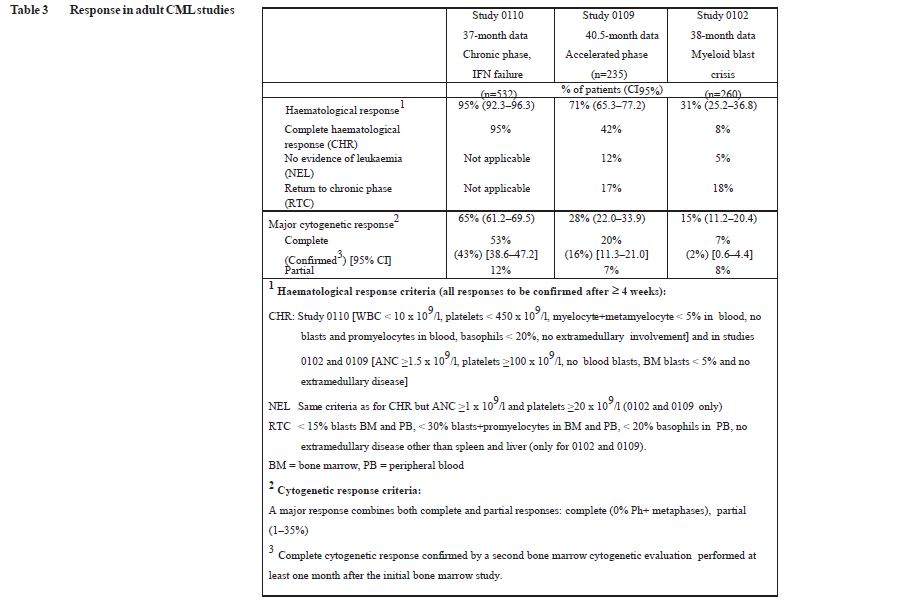
Chronic phase, Interferon failure: 532 adult patients were treated at a starting dose of 400 mg. The patients were distributed in three main categories: haematological failure (29%), cytogenetic failure (35%), or intolerance to interferon (36%). Patients had received a median of 14 months of prior IFN therapy at doses ≥ 25 x 106 IU/week and were all in late chronic phase, with a median time from diagnosis of 32 months. The primary efficacy variable of the study was the rate of major cytogenetic response (complete plus partial response, 0 to 35% Ph+ metaphases in the bone marrow).
In this study 65% of the patients achieved a major cytogenetic response that was complete in 53% (confirmed 43%) of patients (Table 3). A complete haematological response was achieved in 95% of patients.
Accelerated phase: 235 adult patients with accelerated phase disease were enrolled. The first 77 patients were started at 400 mg, the protocol was subsequently amended to allow higher dosing and the remaining 158 patients were started at 600 mg.
The primary efficacy variable was the rate of haematological response, reported as either complete haematological response, no evidence of leukaemia (i.e clearance of blasts from the marrow and the blood, but without a full peripheral blood recovery as for complete responses), or return to chronic phase CML. A confirmed haematological response was achieved in 71.5% of patients (Table 3). Importantly, 27.7% of patients also achieved a major cytogenetic response, which was complete in 20.4% (confirmed 16%) of patients. For the patients treated at 600 mg, the current estimates for median progression-free-survival and overall survival were 22.9 and 42.5 months, respectively.
Myeloid blast crisis: 260 patients with myeloid blast crisis were enrolled. 95 (37%) had received prior chemotherapy for treatment of either accelerated phase or blast crisis (pretreated patients) whereas 165 (63%) had not (“untreated patients”). The first 37 patients were started at 400 mg, the protocol was subsequently amended to allow higher dosing and the remaining 223 patients were started at 600 mg.
The primary efficacy variable was the rate of haematological response, reported as either complete haematological response, no evidence of leukaemia, or return to chronic phase CML using the same criteria as for the study in accelerated phase. In this study, 31% of patients achieved a haematological response (36% in previously untreated patients and 22% in previously treated patients). The rate of response was also higher in the patients treated at 600 mg (33%) as compared to the patients treated at 400 mg (16%, p=0.0220). The current estimate of the median survival of the previously untreated and treated patients was 7.7 and 4.7 months, respectively.
Lymphoid blast crisis: a limited number of patients were enrolled in phase I studies (n=10). The rate of haematological response was 70% with duration of 2-3 months.
Paediatric patients: A total of 26 paediatric patients of age 18 years with either chronic phase CML (n = 11) or CML in blast crisis or Ph+ acute leukaemias (n = 15) were enrolled in a dose-escalation phase This was a population of heavily pretreated patients, as 46% had received prior BMT and 73% a prior multi-agent chemotherapy. Patients were treated at doses of Imatinib of 260 (mg / (m²)) / day(n = 5) 340 mg/m²/day (n=9), 440 mg/m²/day (n = 7) and (570mg / (m²)) / day(n = 5) . Out of 9 patients with chronic phase CML and cytogenetic data available, 4 (44%) and 3 (33%) achieved a partial cytogenetic response, respectively, for rate of MCyR of 77%.
A total of 51 paediatric patients with newly diagnosed and untreated CMLin chronic phase have been enrolled in an open-label, multicentre, single-arm phase II trial. Patients were treated with Imatinib 340 mg/m²/day, with no interruptions in the absence of dose limiting toxicity. Imatinib treatment induces a rapid response in newly diagnosed paediatric CML patients with a CHR of 78% after 8 weeks of therapy. The high rate of CHR is accompanied by the development of a complete cytogenetic response (CCyR) of 65% which is comparable to the results observed in adults. Additionally, partial cytogenetic response (PCyR) was observed in 16% for a MCYR of 81%. The majority of patients who achieved a CCyR developed the CCyR between months 3 and 10 with a median time to response based on the Kaplan-Meier estimate of 5.6 months.The European Medicines Agency has waived the obligation to submit the results of studies with Imatinib in all subsets of the paediatric population in Philadelphia chromosome (ber-abl translocation)-positive chronic myeloid leukaemia.
Clinical studies in Ph+ALL
Newly diagnosed Ph+ALL: In a controlled study (ADE10) of imatinib versus chemotherapy induction in 55 newly diagnosed patients aged 55 years and over, imatinib used as single agent induced a significantly higher rate of complete haematological response than chemotherapy (96.3% vs. 50%; p = 0.0001 ). When salvage therapy with imatinib was administered in patients who did not respond or who responded poorly to chemotherapy, it resulted in 9 patients (81.8%) out of 11 achieving a complete haematological response. This clinical effect was associated with a higher reduction in ber-abl transcripts in the imatinib-treated patients than in the chemotherapy arm after 2 weeks of therapy (p = 0.02) All patients recerved imatinib and consolidation chemotherapy (see Table 4) after induction and the levels of ber-abl transcripts were identical in the two arms at 8 weeks. As expected on of the study design, no difference was observed in remission duration, disease-free survival or overall survival, although patients with complete molecular response and remaming in minimal residual disease had a better outcome in terms of both remission duration (p = 0.01) and disease-free survival (p = 0.02).
The results observed in a population of 211 newly diagnosed Ph+ ALL patients in four uncontrolled clinical studies (AAU02, ADE04, AJP01 and AUS01) are consistent with the results described above. Imatinib in combination with chemotherapy induction (see Table 4) resulted in a complete haematological response rate of 93% (147 out of 158 evaluable patients) and in a major cytogenetic response rate of 90% (19 out of 21 evaluable patients). The complete molecular response rate was 48% (49 out of 102 evaluable patients). Disease-free survival (DFS) and overall survival (OS) constantly exceeded 1 year and were superior to historical control (DFS p < 0.001 OS p < 0.0001 ) in two studies (AJP01 and AUS01).
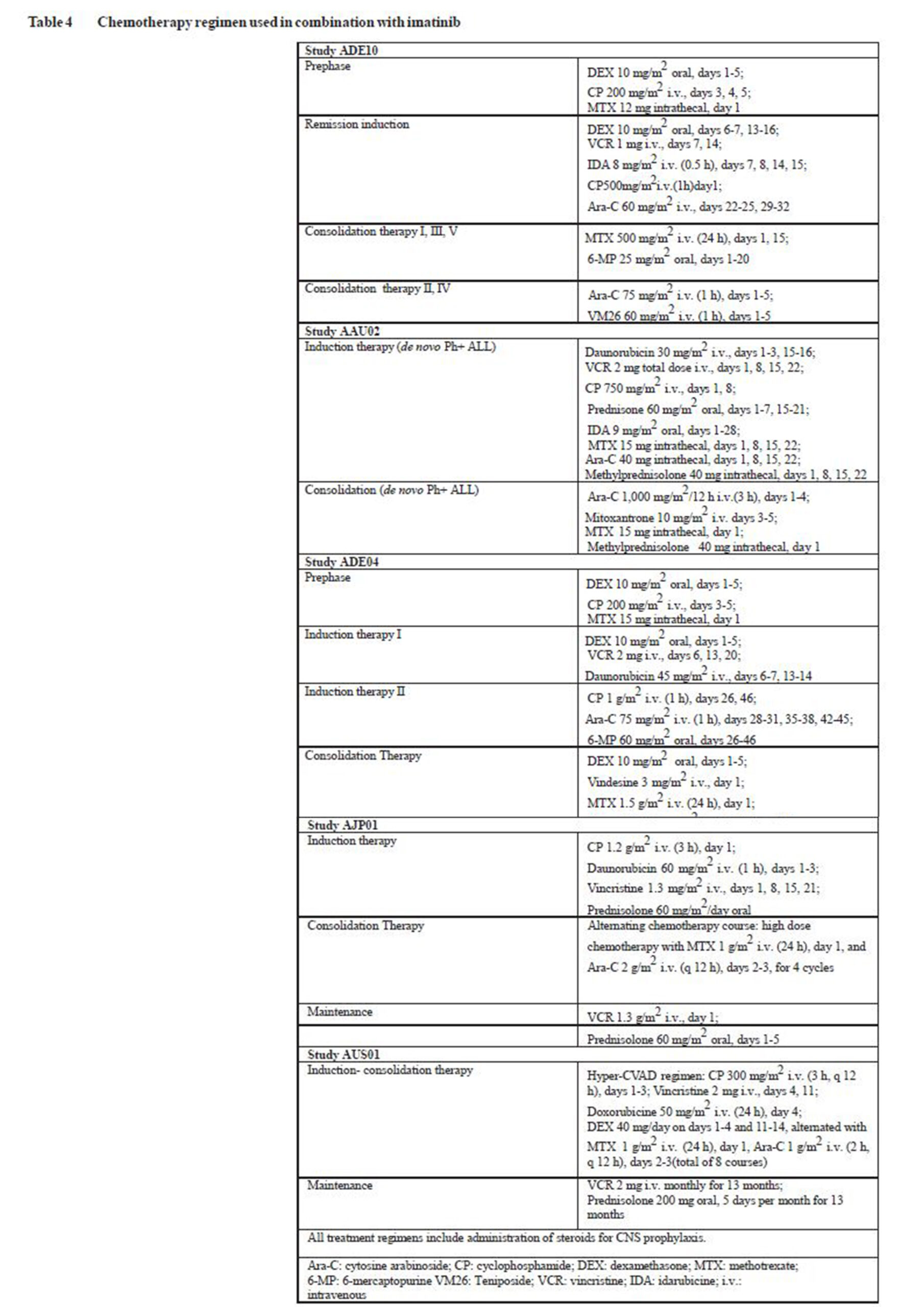
Paediatric patients: In study 12301, a total of 93 paediatric, adolescent and young adult patients (from 1 to 22 years old) with Ph+ALL were enrolled in an open-label, multicentre, sequential cohort, non-randomised phase III trial, and were treated with Imatinib (340 mg/m²/day) in combination with intensive chemotherapy after induction therapy. Imatinib was administered intermittently in cohorts 1-5, with increasing duration and earlier start of Imatinib from cohort to cohort, cohort 1 receiving the lowest intensitiy and cohort 5 receiving the highest intensity of Imatinib (longest duration in days with continuous daily Imatinib dosing during the first chemotherapy treatment courses). Continuous daily exposure to Imatinib early in the course of treatment in combination with chemotherapy in cohort 5-patients (n=50) improved the 4-year event-free survival (EFS) compared to historical controls (n=120), who received standard chemotherapy without Imatinib (69.6% vs. 31.6%, respectively). The estimated 4-year OS in cohort 5-patients was 83.6% compared to 44.8% in the historical controls. 20 out of the 50 (40%) patients in cohort 5 received haematopoietic stem cell transplant.
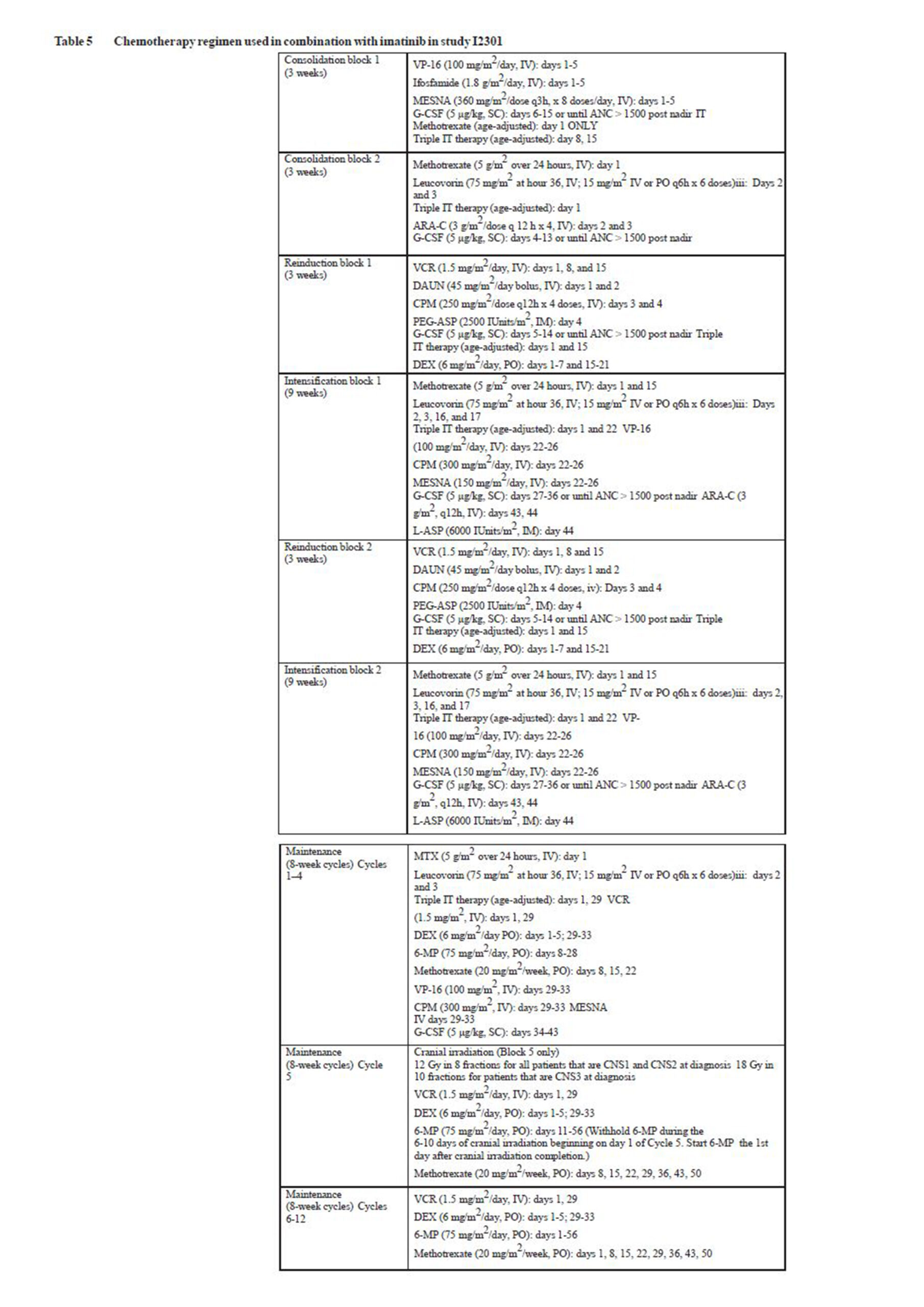
G-CSF granulocyte colony stimulating factor, VP-16 etoposide, MTX methotrexate, IV cytarabine, CPM cyclophosphamide, VCR vincristine, DEX dexamethasone, DAUN asparaginase, MESNA-2-mercaptoethane sulfonate sodium, iii or until MTX level is <0.1 µM, intravenous, SC subcutaneous, IT intrathecal, PO oral, IM intramuscular, ARA-C = daunorubicin, 6-MP 6-mercaptopurine, E.Coli L-ASPL-asparaginase, PEG-ASP PEG q6h=every 6 hours, Gy-Gray
Study AIT07 was a multicentre, open-label, randomised, phase II/III study that included 128 patients (1 to 18 years) treated with imatinib in combination with chemotherapy. Safety data from this study seem to be in line with the safety profile of imatinib in Ph+ ALL patients.
Relapsed/refractory Ph+ ALL: When imatinib was used as single agent in patients with relapsed/refractory Ph+ ALL, it resulted, in the 53 out of 411 patients evaluable for response, in a haematological response rate of 30% (9% complete) and a major cytogenetic response rate of 23%. (Of note, out of the 411 patients, 353 were treated in an expanded access program without primary response data collected.) The median time to progression in the overall population of 411 patients with relapsed/refractory Ph+ALL ranged from 2.6 to 3.1 months, and median overall survival in the 401 evaluable patients ranged from 4.9 to 9 months. The data was similar when re-analysed to include only those patients age 55 or older.
Clinical studies in MDS/MPD
Experience with Imatinib in this indication is very limited and is based on haematological and cytogenetic response rates. There are no controlled trials demonstrating a clinical benefit or increased survival. One open label, multicentre, phase II clinical trial (study B2225) was conducted testing Imatinib in diverse populations of patients suffering from life-threatening diseases associated with Abl, Kit or PDGFR protein tyrosine kinases. This study included 7 patients with MDS/MPD who were treated with Imatinib 400 mg daily. Three patients presented a complete haematological response (CHR) and one patient experienced a partial haematological response (PHR). At the time of the original analysis, three of the four patients with detected PDGFR gene rearrangements developed haematological response (2 CHR and 1 PHR). The age of these patients ranged from 20 to 72 years.
An observational registry (study L2401) was conducted to collect long-term safety and efficacy data in patients suffering from myeloproliferative neoplasms with PDGFR-ẞ rearrangement and who were treated with Imatinib. The 23 patients enrolled in this registry received Imatinib at a median daily dose of 264 mg (range: 100 to 400 mg) for a median duration of 7.2 years (range 0.1 to 12.7 years). Due to the observational nature of this registry, haematologic, cytogenetic and molecular assessment data were available for 22, 9 and 17 of the 23 enrolled patients, respectively. When assuming conservatively that patients with missing data were non-responders, CHR was observed in 20/23 (87%) patients, CCyR in 9/23 (39.1%) patients, and MR in 11/23 (47.8%) patients, respectively. When the response rate is calculated from patients with at least one valid assessment, the response rate for CHR, CČYR and MR was 20/22 (90.9%), 9/9 (100%) and 11/17 (64.7%), respectively.
In addition a further 24 patients with MDS/MPD were reported in 13 publications. 21 patients were treated with Imatinib 400 mg daily, while the other 3 patients received lower doses. In eleven patients PDGFR gene rearrangements was detected, 9 of them achieved a CHR and 1 PHR. The age of these patients ranged from 2 to 79 years. In recent publication updated information from 6 of these 11 patients revealed that all these patients remained in cytogenetic remission (range 32-38 months). The same publication reported long term follow-up data from 12 MDS/MPD patients with PDGFR gene rearrangements (5 patients from study B2225). These patients received Imatinib for a median of 47 months (range 24 days-60 months). In 6 of these patients follow-up now exceeds 4 years. Eleven patients achieved rapid CHR; ten had complete resolution of cytogenetic abnormalities and a decrease or disappearance of fusion transcripts as measured by RT-PCR. Haematological and cytogenetic responses have been sustained for a median of 49 months (range 19-60) and 47 months (range 16-59), respectively. The overall survival is 65 months since diagnosis (range 25-234). Imatinib administration to patients without the genetic translocation generally results in no improvement.
There are no controlled trials in paediatric patients with MDS/MPD. Five (5) patients with MDS/MPD associated with PDGFR gene re-arrangements were reported in 4 publications. The age of these patients ranged from 3 months to 4 years and imatinib was given at dose 50 mg daily or doses ranging from 92.5 to 340 mg/m² daily. All patients achieved complete haematological response, cytogenetic response and/or clinical response.
Clinical studies in HES/CEL
One open-label, multicentre, phase II clinical trial (study B2225) was conducted testing Imatinib in diverse populations of patients suffering from life-threatening diseases associated with Abl, Kit or PDGFR protein tyrosine kinases. In this study, 14 patients with HES/CEL were treated with 100 mg to 1,000 mg of Imatinib daily. A further 162 patients with HES/CEL, reported in 35 published case reports and case series received Imatinib at doses from 75 mg to 800 mg daily. Cytogenetic abnormalities were evaluated in 117 of the total population of 176 patients. In 61 of these 117 patients FIPILI-PDGFRa fusion kinase was identified. An additional four HES patients were found to be FIPIL1-PDGFRa-positive in other 3 published reports. All 65 FIPIL1-PDGFRa fusion kinase positive patients achieved a CHR sustained for months (range from 1 to 44+ months censored at the time of the reporting). As reported in a recent publication 21 of these 65 patients also achieved complete molecular remission with a median follow-up of 28 months (range 13-67 months). The age of these patients ranged from 25 to 72 years. Additionally, improvements in symptomatology and other organ dysfunction abnormalities were reported by the investigators in the case reports. Improvements were reported in cardiac, nervous, skin/subcutaneous tissue, respiratory/thoracic/mediastinal, musculoskeletal/connective tissue/vascular, and gastrointestinal organ systems.
There are no controlled trials in paediatric patients with HES/CEL. Three (3) patients with HES and CEL associated with PDGFR gene re-arrangements were reported in 3 publications. The age of these patients ranged from 2 to 16 years and imatinib was given at dose 300 mg/m² daily or doses ranging from 200 to 400 mg daily. All patients achieved complete haematological response, complete cytogenetic response and/or complete molecular response.
Clinical studies in unresectable and/or metastatic GIST
One phase II, open-label, randomised, uncontrolled multinational study was conducted in patients with unresectable or metastatic malignant gastrointestinal stromal tumours (GIST). In this study 147 patients were enrolled and randomised to receive either 400 mg or 600 mg orally once daily for up to 36 months. These patients ranged in age from 18 to 83 years old and had a pathologic diagnosis of Kit-positive malignant GIST that was unresectable and/or metastatic. Immunohistochemistry was routinely performed with Kit antibody (A-4502, rabbit polyclonal antiserum, 1:100; DAKO Corporation, Carpinteria, CA) according to analysis by an avidin-biotin-peroxidase complex method after antigen retrieval.
The primary evidence of efficacy was based on objective response rates. Tumours were required to be measurable in at least one site of disease, and response characterisation based on Southwestern Oncology Group (SWOG) criteria. Results are provided in Table 6.
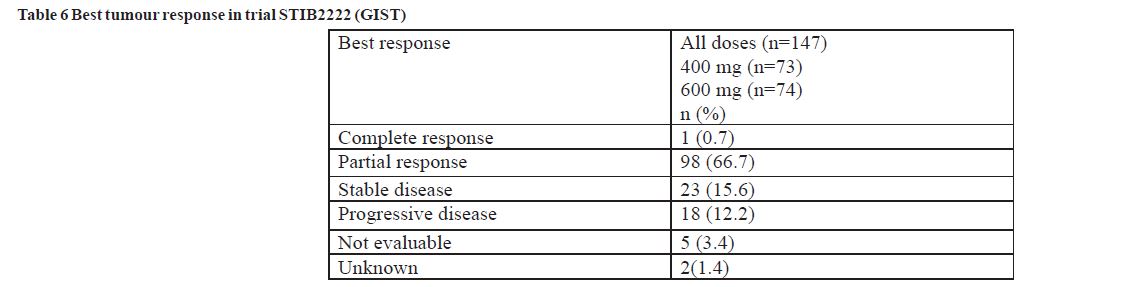
There were no differences in response rates between the two dose groups. A significant number of patients who had stable disease at the time of the interim analysis achieved a partial response with longer treatment (median follow-up 31 months). Median time to response was 13 weeks (95% CI 12-23). Median time to treatment failure in responders was 122 weeks (95% CI 106-147), while in the overall study population it was 84 weeks (95% C.171-109). The median overall survival has not been reached. The Kaplan-Meier estimate for survival after 36-month follow-up is 68%.
In two clinical studies (study B2222 and an intergroup study 50033) the daily dose of Imatinib was escalated to 800 mg in patients progressing at the lower daily doses of 400 mg or 600 mg. The daily dose was escalated to 800 mg in a total of 103 patients, 6 patients achieved a partial response and 21 stabilisation of their disease after dose escalation for an overall clinical benefit of 26%. From the safety data available, escalating the dose to 800 mg daily in patients progressing at lower doses of 400 mg or 600 mg daily does not seem to affect the safety profile of Imatinib.
Pharmacokinetics
Pharmacokinetics of Imatinib
The pharmacokinetics of Imatinib have been evaluated over a dosage range of 25 to 1,000 mg. Plasma pharmacokinetic profiles were analysed on day 1 and on either day 7 or day 28, by which time plasma concentrations had reached steady state
Absorption
Mean absolute bioavailability for Imatinib is 98%. There was high between- patient variability in plasma imatinib AUC levels after an oral dose. When given with a high-fat meal, the rate of absorption of imatinib was minimally reduced (11% decrease in Cmax and prolongation of tmax by 1.5 h), with a small reduction in AUC (7.4%) compared to fasting conditions. The effect of prior gastrointestinal surgery on drug absorption has not been investigated.
Distribution
At clinically relevant concentrations of imatinib, binding to plasma proteins was approximately 95% on the basis of in vitro experiments, mostly to albumin and alpha-acid-glycoprotein, with little binding to lipoprotein.
Biotransformation
The main circulating metabolite in humans is the N-demethylated piperazine derivative, which shows similar in vitro potency to the parent. The plasma AUC for this metabolite was found to be only 16% of the AUC for imatinib. The plasma protein binding of the N-demethylated metabolite is similar to that of the parent compound.
Imatinib and the N-demethyl metabolite together accounted for about 65% of the circulating radioactivity (AUC(0-48h)). The remaining circulating radioactivity consisted of a number of minor metabolites.
The in vitro results showed that CYP3A4 was the major human P450 enzyme catalysing the biotransformation of imatinib. Of a panel of potential comedications (acetaminophen, aciclovir, allopurinol, amphotericin, cytarabine, erythromycin, fluconazole, hydroxyurea, norfloxacin, penicillin V) only erythromycin (IC50 50 µM) and fluconazole (IC50 118 µM) showed inhibition of imatinib metabolism which could have clinical relevance.
Imatinib was shown in vitro to be a competitive inhibitor of marker substrates for CYP2C9, CYP2D6 and CYP3A4/5. Ki values in human liver microsomes were 27, 7.5 and 7.9 µmol/l, respectively. Maximal plasma concentrations of imatinib in patients are 2-4 mol/l, consequently an inhibition of CYP2D6 and/or CYP3A4/5-mediated metabolism of co-administered drugs is possible Imatinib did not interfere with the biotransformation of 5-fluorouracil, but it inhibited paclitaxel metabolism as a result of competitive inhibition of CYP2C8 (Ki = 34.7 µM). This Ki value is far higher than the expected plasma levels of imatinib in patients, consequently no interaction is expected upon co-administration of either 5-fluorouracil or paclitaxel and imatinib.
Elimination
Based on the recovery of compound(s) after an oral C-labelled dose of imatinib, approximately 81% of the dose was recovered within 7 days in faeces (68% of dose) and urine (13% of dose). Unchanged imatinib accounted for 25% of the dose (5% urine, 20% faeces), the remainder being metabolites.
Plasma pharmacokinetics
Following oral administration in healthy volunteers, the t½ was approximately 18h, suggesting that once-daily dosing is appropriate. The increase in mean AUC with increasing dose was linear and dose proportional in the range of 25-1,000 mg imatinib after oral administration. There was no change in the kinetics of imatinib on repeated dosing, and accumulation was 1.5-2.5-fold at steady state when dosed once daily
Pharmacokinetics in GIST patients
In patients with GIST steady-state exposure was 1.5-fold higher than that observed for CML patients for the same dosage (400 mg daily). Based on preliminary population pharmacokinetic analysis in GIST patients, there were three variables (albumin, WBC and bilirubin) found to have a statistically significant relationship with imatinib pharmacokinetics. Decreased values of albumin caused a reduced clearance (CL/f); and higher levels of WBC led to a reduction of CL/f. However, these associations are not sufficiently pronounced to warrant dose adjustment. In this patient population, the presence of hepatic metastases could potentially lead to hepatic insufficiency and reduced metabolism.
Population pharmacokinetics
Based on population pharmacokinetic analysis in CML patients, there was a small effect of age on the volume of distribution (12% increase in patients> 65 years old). This change is not thought to be clinically significant. The effect of bodyweight on the clearance of imatinib is such that for a patient weighing 50 kg the mean clearance is expected to be 8.51/h, while for a patient weighing 100 kg the clearance will rise to 11.8 1/h. These changes are not considered sufficient to warrant dose adjustment based on kg bodyweight. There is no effect of gender on the kinetics of imatinib.
Pharmacokinetics in children
As in adult patients, imatinib was rapidly absorbed after oral administration in paediatric patients in both phase I and phase II studies. Dosing in children at 260 and 340 mg/m2/day achieved the same exposure, respectively, as doses of 400 mg and 600 mg in adult patients. The comparison of AUC(0-24) on day 8 and day 1 at the 340 mg/m2/day dose level revealed a 1.7-fold drug accumulation after repeated once-daily dosing.
Based on pooled population pharmacokinetic analysis in paediatric patients with haematological disorders (CML, Ph+ALL, or other haematological disorders treated with imatinib), clearance of imatinib increases with increasing body surface area (BSA). After correcting for the BSA effect, other demographics such as age, body weight and body mass index did not have clinically significant effects on the exposure of imatinib. The analysis confirmed that exposure of imatinib in paediatric patients receiving 260 mg/m² once daily (not exceeding 400 mg once daily) or 340 mg/m² once daily (not exceeding 600 mg once daily) were similar to those in adult patients who received imatinib 400 mg or 600 mg once daily.
Organ function impairment
Imatinib and its metabolites are not excreted via the kidney to a significant extent. Patients with mild and moderate impairment of renal function appear to have a higher plasma exposure than patients with normal renal function. The increase is approximately 1.5- to 2-fold, corresponding to a 1.5-fold elevation of plasma AGP, to which imatinib binds strongly. The free drug clearance of imatinib is probably similar between patients with renal impairment and those with normal renal function, since renal excretion represents only a minor elimination pathway for imatinib.
Although the results of pharmacokinetic analysis showed that there is considerable inter-subject variation, the mean exposure to imatinib did not increase in patients with varying degrees of liver dysfunction as compared to patients with normal liver function.
PHARMACEUTICAL PARTICULARS
List of Excipients:
Microcrystalline cellulose
(Comprecel M101D+)
Crospovidone
Hypromellose
Iso propyl Alcohol
Microcrystalline cellulose
(Comprecel M102)
Colloidal silicon dioxide
Magnesium stearate
Opadry Yellow (04F52022)
Shelf life: 2 years
Storage: Store in a cool & dry place below 30°C. Protect from light & moisture
Nature and contents of container:
For 400 mg
Blister: Each PVC/PVDC-Alu blister contains 10 Tablets
HDPE: Each HDPE container contains 30 Tablets.
Manufactured By:
M/s Jodas Expoim Pvt. Ltd.
Plot No. 55, Phase III, Biotech Park,
Karkapatla (V), Markook (Mdl),
Siddipet District-502 279, Telangana
INDIA.
Product of:
Grace Biogen Pty Ltd.
Syndney Australia